
How to Select a DNA Sequencing Technology? A Guide to NGS Platforms

Nowadays, there are several sequencing platforms available, each with their unique features that provide alternative options for sequencing mechanisms, read lengths, run times, ease-of-use, and scalability. First-generation platforms are based on classical chain-termination method, colloquially refers to as Sanger sequencing. Second-generation NGS platforms are based on sequencing by synthesis or a pH change detection and generate short reads. These platforms include Ion Torrent devices, sequencers from Illumina, as well as from BGI Group. Third generation NGS platforms can generate long reads, via single molecule real-time or nanopore sequencing. Next, we will review the most used sequencing technologies and their different principles. Understanding the characteristics of each of them is essential to know which one is best suited when designing your studies.
The chain-termination method: Sanger sequencing
Sanger sequencing is a traditional sequencing procedure that uses the chain termination method to generate reads. DNA synthesis is initiated by adding a mixture of deoxynucleotides (dNTPs) along with a small amount of fluorescent-tagged dideoxy nucleotides (ddNTPs). The ddNTPs lacks the 3'-OH group that is required for the formation of a phosphodiester bond with the next nucleotide during DNA synthesis. This causes the synthesis to terminate at the position where the ddNTP is incorporated, resulting in a population of DNA fragments that differ in length by one nucleotide. The fragments are then separated, based on their size, by capillary or gel electrophoresis, and detected by laser excitation of the fluorescent dyes attached to the ddNTPs. Sanger sequencing can generate reads of about 1000 nucleotides in length, with an unprecedented accuracy of 99.9%. Although being the oldest technology, it is still a method of choice, for example in targeted sequencing approaches due to its accuracy, low costs per reaction and fast turnaround times. Validation of variants identified in NGS, or segregation analysis are two examples where sanger sequencing is frequently used.
Semiconductor sequencing
The semiconductor sequencing approach detects nucleotide incorporation during DNA synthesis through ion concentration changes. Ion Torrent is the sequencing platform based on this technology. The platform is essentially a chip that consists of a multi-well plate. Each well contains 5-micron beads, which incorporates around 50,000 amplified DNA fragments. As DNA synthesis proceeds, the incorporation of each nucleotide releases a hydrogen ion. The flow of hydrogen ions causes a change in the pH of the solution that is detected by a sensor at the bottom of the microwell. The nucleotide sequence is then determined by the order of the ion signals.
Ion Torrent sequencing has several advantages over other sequencing technologies. A major advantage of this system is that no camera or light scanner is needed because nucleotide incorporation is directly converted into voltage, which is recorded directly and greatly speeds up the process. It is suitable for targeted sequencing applications, as it allows the sequencing of specific regions of interest (e.g. sequencing of cancer genes, or microbiomes). However, the cost per gigabase (Gb) is relatively high compared to other platforms. Further, it has limitations in read length and accuracy compared to other platforms. The highest throughput chip for the machine can generate 50 Gb of sequence per run, meaning it is not capable of sequencing large regions of DNA or whole genomes, nor detecting structural variant.
Sequencing by synthesis: bridge amplification
Sequencing by synthesis technology uses a polymerase or ligase enzyme to incorporate nucleotides with a fluorescent tag into the sequence, which are then identified to determine the DNA sequence. Before sequence analysis, an amplification step is needed to generate a large enough number of copies of each DNA template so that there is sufficient signal strength for each base addition. On of the most common techniques used to amplify DNA is the bridge amplification, utilize by Illumina sequencing platforms.
The Illumina sequencing workflow involves several steps, including library preparation, cluster generation, sequencing, and data analysis. During library preparation, DNA is fragmented into small pieces and adapters are added to both ends of the fragments to facilitate their binding to a flow cell. The flow cell contains millions of small wells, or clusters, where individual DNA fragments are amplified through bridge amplification generating clusters of identical DNA fragments. After cluster generation, the sequencing process begins. In each cycle of sequencing, a nucleotide base is added to the growing DNA strand, and a fluorescent signal is emitted that corresponds to the incorporated base. The fluorescent signal is recorded, and the base call is made depending on the color and intensity of the signal.
Illumina sequencing typically generates reads that range in length from 50 to 600 base pairs, with the most used read length being 300 base pairs (paired end). The length of the read depends on the specific Illumina sequencing instrument and the application for which the sequencing is being performed. One of the key advantages of Illumina sequencing is its high accuracy, with an error rate of less than 1% per base. Additionally, it is highly scalable, allowing for the sequencing of large or small genomes, transcriptomes, and metagenomes. This makes it a versatile technology with many applications, including whole genome sequencing, transcriptome profiling, epigenetic analysis, and metagenomics.
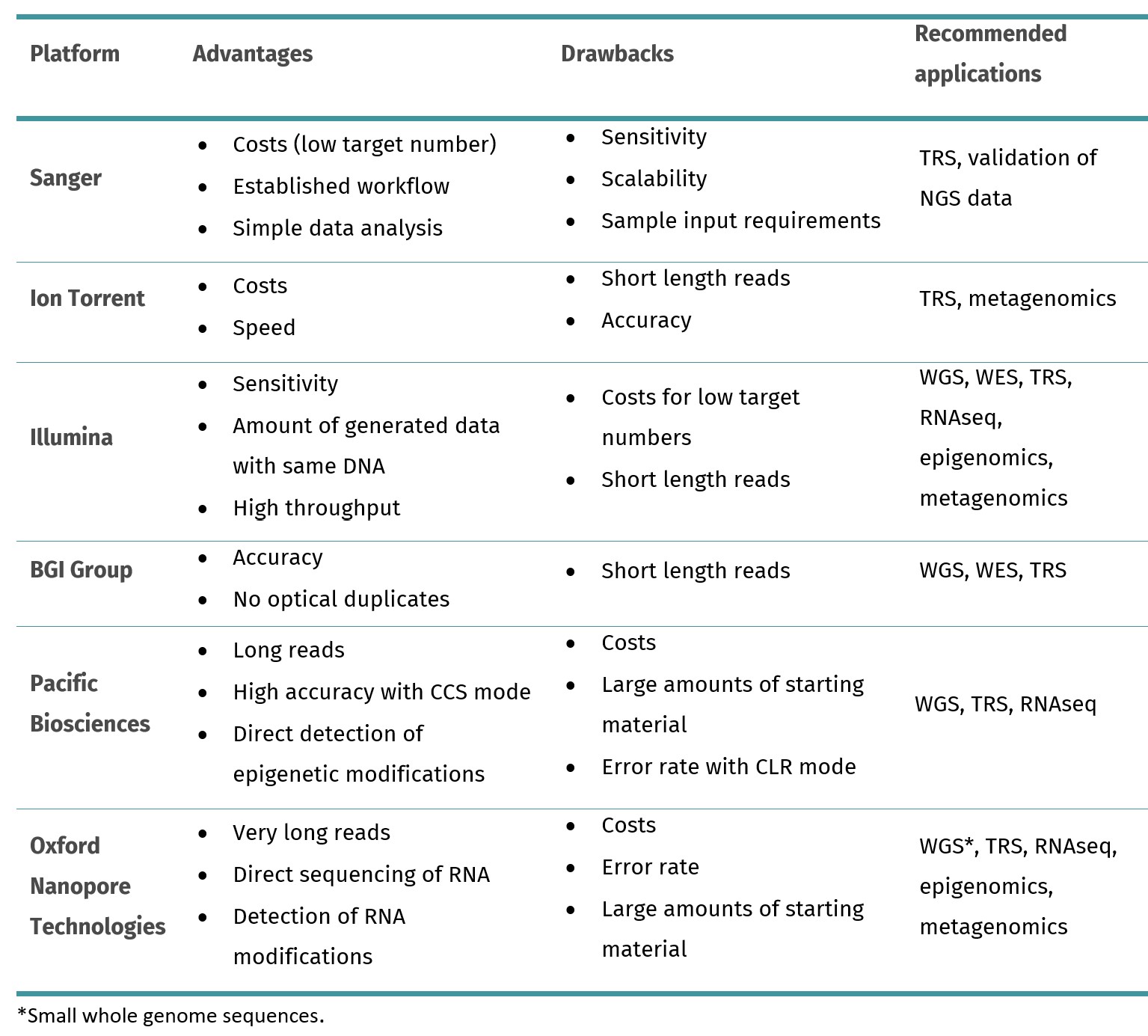
Table 1. Overview of the most common sequencing platforms. WGS: whole genome sequencing; WES: whole exome sequencing; TRS: targeted sequencing, RNAseq: RNA sequencing; CCS: circular consensus sequencing; CLR: continuous long read sequencing.
Combinatorial probe-anchor synthesis: nanoball sequencing
This sequencing technique was developed by the BGI Group (formerly known as Beijing Genomics Institute). The so-called combinatorial probe-anchor synthesis (cPAS) consists of rolling circle replication with the Phi 29 DNA polymerase, which synthesizes a long, single-stranded DNA that self assembles into a nanoball (around 300 nm across). Fluorescent probes are incorporated, and the nanoballs are attached to a silicon wafer flow cell where they selectively bind to the positively charged material in a highly ordered pattern. The emission of fluorescence is then imaged and measured to record the base position.
Common to all short-read sequencing techniques, the main disadvantage is that you cannot obtain long lengths of DNA sequences. Nevertheless, an important advantage of cPAS-based sequencing is that the high accuracy of Phi 29 DNA polymerase ensures accurate amplification of the circular template. Also, because the DNA nanoballs remain in place on the flow cell, they do not produce optical duplicates and do not interfere with neighboring DNA.
Single molecule real-time sequencing (SMRT)
SMRT sequencing generates long reads with high accuracy, being the core technology that drives Pacific Biosciences (PacBio) platforms. The workflow begins with the preparation of a single-stranded circular template DNA molecule that is immobilized on a zero-mode waveguide (ZMW) array. The ZMW array is a tiny well that is designed to allow for the observation of individual DNA molecules. The DNA polymerase enzyme is then added to the well, along with fluorescently labeled nucleotides, which are incorporated into the growing DNA strand. As the DNA polymerase extends the template strand, the fluorescently labeled nucleotides are incorporated into the DNA molecule, and the resulting signal is recorded by a detector. The fluorescent signal is then used to determine the identity of the nucleotide that was incorporated, allowing for the generation of a long read. You can optimize your results with two sequencing modes: the circular consensus sequencing (CCS), which produces highly accurate long reads, and the continuous long read (CLR) sequencing to generate the longest possible reads.
The SMRT sequencing technology is unique in that it can generate reads that are tens of kilobases in length. A problem that PacBio faces is that the platform only sequences individual molecules, and the signal strength of base incorporations is quite low. Although the zero-mode waveguides measure some signals, the DNA polymerase adds bases so quickly that the ability to detect growing sequences is challenging. However, these sequencing errors are random, meaning that if the same fragment is sequenced multiple times, a higher overall accuracy can be achieved. Historically, it has been associated with higher error rates, but that is changing, and SMRT sequencing technology enables an overall sequencing accuracy of 99%, although this results in a slightly lower output. Overall, it is a powerful technology that enables the generation of long reads with high accuracy, making it a valuable tool for a variety of applications including de novo genome assembly, targeted sequencing, and transcriptome analysis.
Nanopore sequencing
Oxford Nanopore Technologies provides a nanopore-based approach to directly sequence DNA or RNA molecules in real-time. The nanopore sequencing workflow begins with the preparation of a single-stranded DNA or RNA molecule that is passed through a protein nanopore embedded in a membrane. The nanopore is designed to allow individual nucleotides to pass through the pore, which results in a change in electrical current across the membrane. This change in current is detected and processed by a computer in real-time to determine the identity of each nucleotide.
One of the unique features of nanopore sequencing is the ability to generate reads that are several kilobases in length, which is longer than other sequencing technologies. This feature makes it possible to assemble complex genomes with high accuracy and to identify structural variants in the genome. Another advantage of nanopore sequencing is the ability to detect modifications in the DNA or RNA molecule, such as methylation and base modifications. This is accomplished by using specific nanopores that are designed to recognize these modifications, which results in a unique electrical signal. Oxford Nanopore sequencing also offers the advantage of being portable and real-time, with a small device (MinION) allowing for sequencing in the field or in remote locations. Despite its advantages, nanopore sequencing has some limitations, including a relatively high error rate compared to other sequencing technologies. However, ongoing developments in technology and bioinformatics tools are expected to improve the accuracy and reliability of nanopore sequencing in the future.
Considerations for your sequencing: which platform fits better?
When choosing a sequencing platform, you should consider several factors, particularly with respect to the downstream application or the specific research goals. If your aim is to sequence the entire genome of an organism (WGS), considerations include the sequencing platform's read length, accuracy, coverage depth, and cost per base, as WGS generates a large volume of data. We recommend platforms with high throughput and accuracy. When your study focusses on protein-coding regions of the genome (WES), we prefer platforms with high sequencing depth and accuracy, for capturing exonic regions effectively. Target sequencing (TRS) focuses on specific genomic regions and needs high sequencing depth to detect variants. You should also evaluate the required read length for the targeted regions to select a platform that meets the specific needs of these regions. For transcriptomics studies (RNA-seq), the important factors to consider include the sequencing platform's sensitivity, dynamic range, and ability to detect alternative splicing. Epigenomic studies focus on DNA modifications that need sequencing platforms with the ability to detect these modifications. Long-read sequencing technologies are advantageous for mapping modified regions accurately, being preferred the nanopore sequencing. Microbiome sequencing will require sequence throughput and sufficient accuracy to differentiate between closely related species. Illumina sequencing is widely used for microbial studies due to its high throughput and accuracy.
In addition, nucleic acid sample requirements play an essential role during the selection of an application and a platform, which will be determined at the same time by your sample input type and the extraction method you choose. If you want to use one of the third generation NGS platforms, you should know that those require fresh DNA for ensuring quality of ultralong reads. Regarding different applications, WES generally requires lower DNA amount compared to WGS. For epigenomics analysis, as well as WGS and TRS applications, you will need a high-quality DNA and a quantity ranging from several micrograms to hundreds of nanograms, depending on the sequencing platform. RNA-seq requires high-quality total RNA or mRNA samples, and the amount of RNA required can range from micrograms to tens of nanograms, depending on the platform and desired coverage depth. Your RNA should be of good quality with high integrity to ensure accurate representation of gene expression profiles.
BioEcho provides a range of kits for DNA and RNA extraction, which delivers high-quality products ready-to-use in your downstream applications. The quality of your nucleic acids determines the success of your sequencing. We advise consulting the manufacturer's guidelines and specific experimental protocols for each application to ensure that optimal sample requirements are met. Discover our products and talk to our nucleic acid experts about your projects to better understand your necessities.

Laura is a passionate scientific communicator, with an extensive experience as a researcher in several fields, including human genetics, biotechnology, and neuroscience. Since joining BioEcho in 2022, she enjoys creating appealing content and material for our customers and interested parties. Laura likes practicing yoga, cooking wholesome Mediterranean food, and playing the piano. She also loves spending weekends on the nature, rock climbing and hiking.